Mopathic - Diagnose av alvorlig. I denne artikkelen forteller utøveren om hvilke typer av denne mangesidede sykdommen.
Innhold
 «Vi er tre brødre (31 år gammel, 29 og 27 år) - Med ungdomsår lider jeg av en sykdom - Progressiv Muscle Dystrofi. Alle tre - funksjonshemmede. Kanskje spesialister vil bli tilbakebetalt på vår ulykke og hjelp.»
«Vi er tre brødre (31 år gammel, 29 og 27 år) - Med ungdomsår lider jeg av en sykdom - Progressiv Muscle Dystrofi. Alle tre - funksjonshemmede. Kanskje spesialister vil bli tilbakebetalt på vår ulykke og hjelp.»
«I et barn (10 år gammel), den progressive muskeldystrofi-disen. Legene maktesløs. Jeg vil være takknemlig for eventuelle oppskrifter, tips.»
«Barnebarnet (3 år) begynte plutselig å nekte ben og over tid det blir verre. Legene legger forskjellige diagnoser og kan ikke gjøre noe. Hjelp gode råd.»
Disse er utdrag fra brev av mennesker som kolliderte med en så alvorlig sykdom som myopati. Hva er myopati? La oss prøve å klassifisere dette multi-diagrammet.
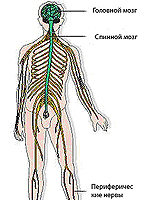
Myopati og dets typer
Moopathi representerer en gruppe nevromuskulære sykdommer som manifesterer seg tretthet, muskelsvakhet, en reduksjon i muskeltonen, muskelatrofi. Moopathy, avhengig av årsakssammenheng, er delt inn i progressiv arvelig muskeldystrofi, endokrin myopati (sykdommer i de interne sekresjonskjertlene) og metabolske myopati (metabolske lidelser).
Snakk om den progressive arvelige muskeldystrofi. Denne typen myopati er preget av muskelatrofi på grunn av ødeleggelsen av muskelceller på grunn av en ulempe ved et spesielt protein, som styrker strukturen av muskelfibre. Dette proteinet fremstilles under kontroll av et spesielt cellegen, som ligger på det 6. humane kromosom, og under defekten av dette genet kommer den gradvise ødeleggelsen av muskelcelleskall, etterfulgt av støping av muskelfibre.
Dette defekte genet er arvet hvis det var ekteskap mellom slektninger. Geneendringer i 30% av tilfellene oppstår som følge av mutasjon, det vil si i disse tilfellene ekteskap mellom slektninger - verken. Sykdommen er arvet med en 50% sannsynlighet, hvis en av foreldrene til barnet er syk. Det er forbundet med det kvinnelige sexkromosomet og overføres som regel sønner, selv om kvinner selv ikke kan skade. Atrofi Musklene i skulderbeltet, tilbake, bekkenbeltet og bena.
Avhengig av lokaliseringen av sykdommen, alder, alder, alvorlighetsgraden av sykdommene tildeler ulike former for muskeldystrofi. Så, ungdomsdannelsen av Erba Rota forekommer i en alder av 10-20 år, da atmosfæren i skulderbeltet og hendene virker umerkelig, og deretter - bekkenbeltet og bena. Mens du går på pasienten, magen og vridd baksiden av brystet. For å skille seg ut av stillingen som ligger, slår pasienten på hans side og lener hendene på hoftene, gradvis øker kroppen sin. Sykdommen utvikler seg sakte.
Barns form for muskeldystrofi av DCUSCEA begynner i en alder av 3-5 år med atrofi av musklene bekken, hofter med samtidig fortykkelse av oscraconal muskler i beinet (falsk fortykkelse). Gradvis atrofi muskler av skulder belter og hender. Barn opprinnelig forstyrret gang, og deretter oppstår vanskelighetsgrad i bevegelse. Mange har en hjertefrekvens på grunn av en økning i hjerteslag. Progresjonen av sykdommen eller dens ondartede strømning på grunn av tidlig immobilisering av lemmer fører til et trist utfall. De er syke, for det meste gutter (1 for 3000 født). For å være mer nøyaktig, er mennene og kvinnene også syke. Bare Doses sykdom manifesterer seg i gutter. Jenter er bærere av dette genet.
Men det skjer og godartet for muskelsdystrofi (Becker Miodastrofi), når sykdommen manifesterer seg sakte, spesielt i lavspennende barn. I mange år beholder de en tilfredsstillende fysisk tilstand, og bare tiltredelsen av ulike akutte sykdommer og skader fører dem til immobilisering, utmattelse med dårlig utfall.
Den skulder-slag-ansiktsform for Miodyatrofofi, kalt Landuzy-Dezhard, som kan være i alderen 6 til 52 år (oftere i 10-15 år) og er preget av nederlaget av musklene i ansiktet med gradvis påfølgende atrofi av musklene i skulderbeltet, torso og lemmer. Tidlige tegn på sykdommen er dårlig lukkede og uklare øyelokk, fullt lukkede lepper, som skaper en fuzzy tale og umuligheten av oppblåsende kinn. Sykdommen fortsetter sakte. I lang tid kan pasienten bevege seg og opprettholde evnen til å jobbe, og deretter etter 15-25 år blir musklene i bekkenbeltet på bena gradvis atrofisert, noe som gjør det vanskelig å bevege seg.
En gruppe sekundære progressive muskeldystrofi er også tildelt, som oppstår i forbindelse med skadenes skade: Neural, Spinal Miodastrophia, kalt Still Amyotrofi.
Shark-Marie Amyotrofi-bilde, som er preget av gradvis atrofi av de små musklene i stoppet, blir deretter atrofiens muskler i bena og den nedre delen av hoftene, og musklene i midten og øvre deler av hoftene ikke gjør det endring og låret er formen på en flaske med nakke, vippet ned. Musklene i hendene og underarmene blir så gradvis atrofi. Muskler i torso, skulderbelte og ansikt. Sykdommen oppstår i en alder av 18-25, går langsomt og stabiliserer seg.
Medfødt spinalmuskulær atrofi av Kugelberg-Vallanger er preget av gradvis atrofi av musklene i hendene, bena, retardasjon av mental og fysisk utvikling, deformasjon av ryggraden. Sykdommen manifesteres i en alder av 8-10 år og går langsomt.
Den progressive ayotroofonien i Aran-Duzhen begynner i en alder av 25-50 år og manifesterer muskelatrofien av børstene. Så er resten av hendene gradvis atrofisert, deretter kroppens føtter, i t.C. intercostal muskler, som forårsaker respiratoriske lidelser fra hvilken død kommer.
Medfødt amiotonia (reduksjon av muskeltonen) Oppenheim er preget av svakheten i musklene på grunn av deres underutvikling, og deres muskeldystrofi er sekundær. I det nyfødte utvikler det ikke, men inngangen av respiratoriske infeksjoner kan forårsake betennelse og døden kommer i det første året av livet. Med alderen forbedrer muskelmotorfunksjonen.
Behandling av muskelsdystrofi er rettet mot å bremse de dystrofiske (ødeleggende) prosessene i musklene og til og med deres oppsigelse. Imidlertid har radikalbehandling ennå ikke blitt funnet. Selv om håp er på geninterapien, som begynner å sakte implementere i medisinsk praksis.